USHER BRIGHTNESS ist ein "Aufruf zur Aktion", die Forschung am erblich bedingten, noch unheilbaren Usher-Syndrom mit Spenden zu fördern. Dazu verweise ich auf die deutsche Stiftung PRO RETINA und die kanadische Foundation Fighting Blindness.
Was wäre schlimmer – taub oder blind zu sein?
Diese Frage haben sich bestimmt ein paar von Euch bereits gestellt. Stellt Euch nun vor, taub und blind zugleich zu sein. Für Betroffene ist das zunächst sehr schlimm. So auch für mich (von USH2A betroffen). Mich hat der Gedanke, eines Tages möglicherweise zu erblinden, lange gequält. Abhängigkeit von fürsorglichen Personen, die mir rund um die Uhr helfen, droht, dachte ich. Ein Gefühl von Hilflosigkeit.
Ich habe mich arrangiert. Komme heutzutage mit der starken Hör- und Seheinschränkung klar. Dennoch lässt mich der Gedanke nicht los, eines Tages einwandfrei im Dunklen unterwegs zu sein, sicher immer Autofahren zu dürfen, auch im Alter neue optische (v.a. räumliches Sehen, Farbensehen) und akustische Eindrücke im vollen Ausmaß genießen zu dürfen. Geschweige denn, meinen Alltag überhaupt unabhängig beschreiten zu können.
Das Usher-Syndrom ist zwar nicht lebensbedrohlich, dennoch nimmt es im Verlauf der Krankheit sehr viel an Lebensqualität.
Usher Syndrom


Kurze Erklärung
Das Usher-Syndrom ist benannt nach dem britischen Augenarzt Charles Usher, der 1914 die Vererbung dieser Erkrankung beschrieb.
Es ist eine autosomal-rezessiv vererbte Erkrankung, die durch die Kombination einer Schwerhörigkeit/Taubheit mit einer Sehstörung in Form der Auswirkungen einer Retinopathia Pigmentosa charakterisiert ist.
Es gibt ca. 40 bekannte Syndrome, die Gehörlosigkeit und Blindheit als Symptome beinhalten. Das Usher-Syndrom ist mit ca. 100.000 bekannten Fällen der Gesamtbevölkerung die häufigste Form angeborener Taubblindheit des Menschen.
Verlauf
Der Verlauf der Krankheit wird als mittel- bis hochgradige Innenohrschwerhörigkeit/Gehörlosigkeit von Geburt an verbunden mit einem später einsetzenden, fortschreitenden Sehverlust beschrieben.
Der Sehverlust wird durch die sog. Retinopathia Pigmentosa (RP;früher “Retinitis pigmentosa”) verursacht und beginnt im äußeren Bereich. Die stäbchenförmigen Sehzellen sterben ab. Dies hat zur Folge, dass der Betroffene Nachtblind wird. Im weiteren Verlauf verengt sich das Gesichtsfeld zu einem Tunnelblick, das Farbsehen läßt nach und es tritt eine gesteigerte Blendungsempfindlichkeit auf. Die Sehschärfe bleibt relativ lange erhalten. Der Sehverlust ist häufig schleichend und individuell unterschiedlich und kann bis zur Erblindung führen.




Die vorhandene Hörbeeinträchtigung (Taubheit oder mittel-bis hochgradige Schwerhörigkeit) wird im Wesentlichen durch eine Schädigung der Haarzellen in der Innenohr-Schnecke verursacht.
Subtypen
Es gibt grob gesehen je nach Schwerhörigkeit drei Typen von Krankheitsbildern des Usher-Syndroms:
Typ I (USH1) Angeborene Gehörlosigkeit in Kombination mit im Kindesalter beginnender RP; zusätzlich in vielen Fällen Gleichgewichtsstörungen (Untergruppen A-G, am häufigsten Usher I B)
Typ II (USH2) Unterschiedlich ausgeprägte (mittel- bis hochgradig), relativ konstant bleibende Schwerhörigkeit in Kombination mit einer in der Pubertät einsetzenden RP (Untergruppen A-C, II A als häufigste Form des Usher-Syndroms)
Typ III (USH3) Fortschreitender Hörverlust im späteren Erwachsenenalter (postlingual) in Kombination mit einer zum ähnlichen Zeitpunkt einsetzenden RP; sehr selten, bisher nur in Finnland und den USA nachgewiesen
Ursachen
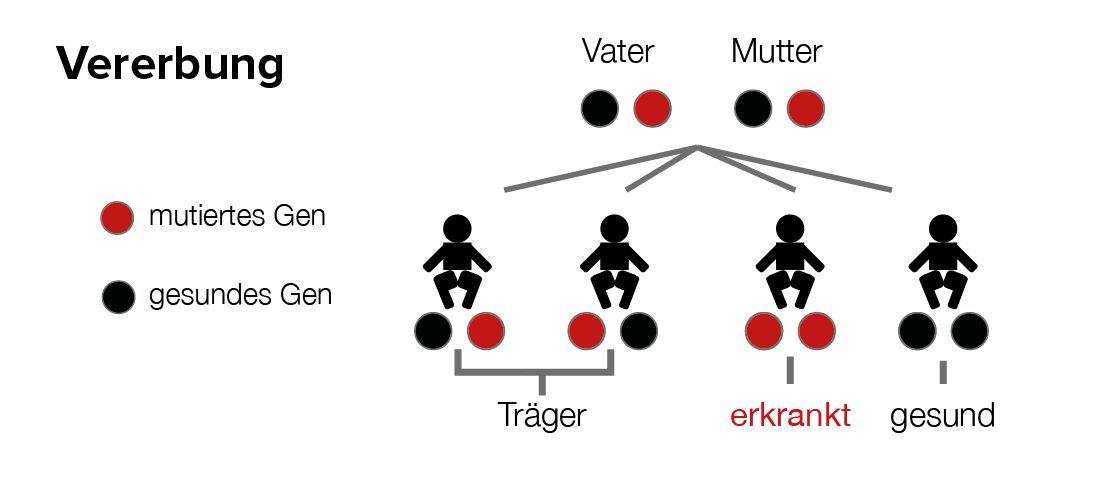
Ursache für die unterschiedlichen Subtypen der Krankheit sind mehrere unabhängige Loci auf verschiedenen Chromosomen, in denen das Usher-Syndrom verursachende Gendefekte entdeckt wurden.
Das Usher-Syndrom kommt im Vergleich zur reinen RP selten vor. Dies erklärt sich aus dem autosomal-rezessiven Erbgang. Im Falle, dass der Betroffene je ein für das Usher-Syndrom verantwortliches, defektes Gen von Mutter und Vater erhalten hat, kommt es mit einer nur 25%igen Wahrscheinlichkeit zur Erkrankung des Kindes am Usher-Syndrom. Kein Elternteil ist selbst davon betroffen.
Defekte Genorte sind natürlich die Hauptindizien für die Kombination aus Netzhautdegeneration und Innenohrschwerhörigkeit.Der Verlauf der Krankheit wird allerdings auch von sog. „Hintergrundgenen“ (= nicht direkt Usher-Gene), Umwelteinflüssen und psychischen Faktoren beeinflusst.
Immer mehr Ursachen für das Usher-Syndrom werden mit der Zeit aufgedeckt, weshalb die Krankheit hoffentlich in Zukunft besser diagnostiziert werden kann. Erst dann sind Therapieversuche denkbar.
Behandlung und Forschung
Während der Gehörverlust mit Hörgeräten und Cochlea-Implantaten ausgeglichen werden kann, gibt es bislang noch keine Therapiemöglichkeit für das Auge.
Forscher gehen zunächst weiter den Ursachen der Krankheit auf den Grund. Nur so konnten sie feststellen, dass das Usher–Syndrom verschiedene, voneinander unabhängige Subtypen beinhaltet. Fortschritte hinsichtlich der Aufdeckung genetischer Ursachen für das Usher Syndrom geben Hoffnung auf bessere Diagnostik und eventuelle Therapieansätze. Innerhalb der letzten Jahre konnten neun Usher-Gene (Subtypen) identifiziert werden.
Eine wirkliche Heilmethode wäre die Gentherapie. Man versucht, durch das Ersetzen veränderter Krankheitsgene mit Intakten zu verhindern, dass die Krankheit fortschreitet oder überhaupt ausbricht. Die Anwendung beschränkt sich bislang jedoch auf Tierversuche, weil sie am Menschen noch nicht verantwortbar ist.
Neben der Gentherapie gibt es weitere Therapien, die jedoch keine Heilung, sondern mehr oder weniger nur auf eine Verlangsamung der Netzhauterkrankung (RP) hinauslaufen. Transplantationen gelangen bisher nur für einige Zelltypen der Netzhaut. Hierbei liegt die Hoffnung bei der Transplantation von Stammzellen.
Des Weiteren liegen für Nahrungsergänzungsmittel wie Vitamin A, DHA oder Zeaxanthin zwar positive, aber nicht überzeugende Hinweise vor, einen Einfluss auf den Verlauf der Sehschädigung zu haben. Mit einem besonders vielversprechenden Ergebnis schnitt bei langjährigen Untersuchungen durch Prof. Berson das Vitamin A ab. Es erwies sich, dass bei Patienten ein 20% verringertes Fortschreiten der RP auftrat, sofern sie täglich eine Dosis von 15.000 internationalen Einheiten (I.E.) Vitamin-A-Palmitat (Retinyl-Palminat) einnahmen. Erhöhte Leberwerte können allerdings als Nebenwirkung auftreten.
Wie sich herausstellt, gibt es keine sehr wirksamen Behandlungen und erst recht kaum Ansätze für Heilmethoden. Dennoch schenken diese wenigen Therapieansätze heutzutage Hoffnung, wenn man den Stand der Dinge um 1858 in Betracht zieht. Zu dieser Zeit erfolgte durch Albrecht von Graefe (1828-1870), den Begründer der modernen Augenheilkunde, die Ersterwähnung der Kopplung von Seh- und Hörbehinderung, in der es unter anderem lautet:
„Der so ausserordentlich langsame, aber wie es scheint gleichförmige Verlauf dieser Krankheit, das Wesen der anatomischen Veränderung, nämlich die innige Verbindung der Pigmentablagerung mit der Nerven-Atrophie, die circumscripte Abgränzung eines noch gut erhaltenen Gesichtsfeldabschnittes gegen den Defect und endlich das häufige hereditäre Vorkommen, das Alles bestätigt wohl zur Zeit die Ansicht, dass es sich hier um eine tiefwurzelnde trophische Störung handelt, gegen welche wahrscheinlich die Therapie für alle Zeit ohnmächtig bleiben wird.“
Basierend auf heutigen Therapie-Ideen bzw. -Ansätzen sollten Therapiemöglichkeiten hoffentlich nicht „für alle Zeit ohnmächtig bleiben“. Wissenschaftlich fundamentale Entdeckungen innerhalb der letzten Jahre sind „Lichtblicke“.
Ihr könnt dazu beitragen, dass das Usher–Syndrom schnellstmöglich heilbar sein wird.
Jeder einzelne Spendenbeitrag von Euch zählt. DANKE!