USHER BRIGHTNESS is a "call to action" to support the research of the inherited, still incurable Usher syndrome with donations. Therefore I give reference to the German Foundation PRO RETINA and the Canadian Foundation Fighting Blindness.
What would be worse - to be deaf or blind?
Some of you might have asked yourself this question: How would it be if you were deaf and blind at the same time?
For the person affected, this possibility is very threatening...
...as it was for me (affected by USH2A). The thought that one day I might go blind has been bothering me for a long time. Being dependent on the help of caring people around the clock… seemed threatening to me. This gave me a pure feeling of helplessness.
I came to an acceptance. Today I have found a way to live with this disease. However, the thought of one day being able to be on the road in the dark, to safely travel by car, and at old age being able to enjoy acoustic and visual impressions with the help of new optical (spatial and color vision) and acoustical development, is what I hope for - let alone the wish and determination to lead an independent, autonomous life.
Although the Usher syndrome is not life threatening, the progress of the disease gradually reduces quality of life.
Usher Syndrome

Usher Syndrome derives its name from the British opthomologist Charles Usher, who, in 1914, was able to define the disease. The condition is an autosomal recessive inherited disease, characterized by both a hearing disability/deafness and a visual disability, in the form of a Retinopathia Pigmentosa.
There are approximately 40 known Syndromes that bear deafness and blindness as symptoms. With 100,000 known affected individuals globally, Usher Syndrome is the most common hereditary deafness/blindness disease.
Process
The process of the illness is defined as a medium-to high level hearing impairedness/deafness starting at birth, in combination with a subsequent progression loss of sight.
The loss of sight is caused by the so-called Retinopathia Pigmentosa (“RP“; formerly known as “Retinitis Pigmentosa“), and begins at the outer area. The rod-shaped visual cells die off. The result is that the affected person suffers from night blindness. During the continuous progression, the field of vision narrows down to a tunnel vision.
Further, defining color becomes more difficult and there is an increased sensitivity to glare. The visual acuity remains preserved for a relatively long time. The loss of sight is frequently gradual and is different from individual to individual, possibly leading up to complete blindness in some cases.




The existing hearing impairment (deafness or a middle-to-high-grade hearing disability) is primarily caused by the damage of hair cells in the innerear.
Types
Based on the level of hearing impairment, there are three categories that the illness can be separated into:
Usher Type I (USH1): Deafness at birth in combination with the onset of RP in childhood; further, in many of the cases, the individual has a problem with his/her balance. (Sub-group A-G, most frequently Usher I B)
Usher Type II (USH2): defined by various levels (middle-to-high-grade), relatively constant remaining hearing impairment, in combination with the onset of RP at puberty (Sub-group A-C, II A as the most frequent form of the Usher Syndrome)
Usher Type III (USH3): Progressive loss of hearing at later-adulthood (postlingual), in combination with an onset of RP at about the same time; very seldom, with known cases only in Finland and the USA (Sub-group A and B)
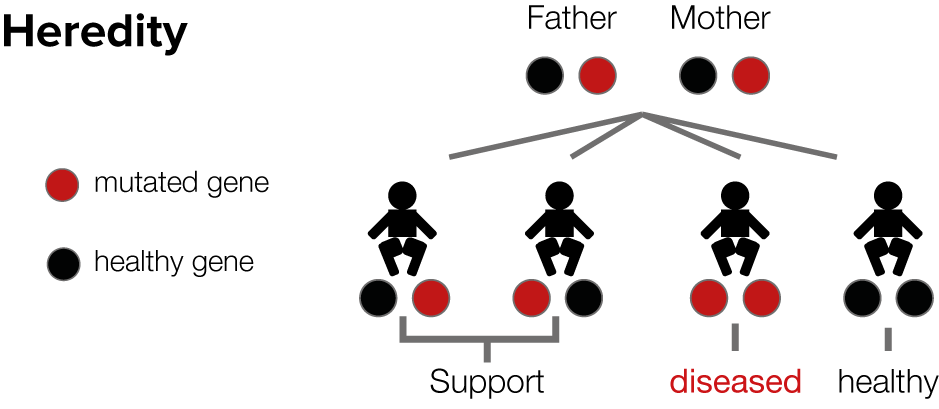
Usher syndrome occurs very rarely compared to retinitis pigmentosa (RP) because of its autosomal recessive inheritance. If the person affected has obtained the defective genes responsible for Usher syndrome from both the mother and father, there is a 25% chance that the child will have Usher syndrome. The parents themselves are not affected.
Defective gene loci are the main evidence for the combination of retinal degeneration and inner ear hearing loss. However, the process of the disease is also influenced by so-called “background genes“ (no actual Usher genes), environmental influences, and emotional factors. Gradually, over the course of time, more causes for Usher syndrome are being discovered, so hopefully this disease can be diagnosed more successfully in the future. Only then can more successful treatment methods be developed.
Research and Treatment
While hearing loss can be improved with the use of hearing aids and cochlea implants, there have not been any therapies discovered for the eye yet. Researchers are still working on the causes of Usher syndrome and have been able to identify different, independent subtypes of Usher syndrome. Progress in revealing genetic causes has raised hope of better diagnostics and possible therapeutic approaches. Within the last couple of years, nine new Usher subtypes could be identified.
Actually gene therapy would be the best treatment. Mutated genes are being substituted with healthy genes in order to prevent the disease from progressing or even breaking out. So far, practical application has been restricted to animal testing, as experiments on human beings cannot be conducted yet.
Other therapies can slow down the progress of RP, but cannot cure the disease. Retina transplantations have only been successful with some cell types of the retina. Dietary supplements with vitamin A, DHA or zeaxanthin show positive but not convincing influence on the delay of sight impairment. Long-term studies by professor Berson showed promising results using vitamin A with patients taking a daily dose of 15,000 units of vitamin A palmitat (retinylpalmitat). However, a negative side effect can be increased liver function reading. Hope lies with stem cell therapy.
So far, there does not exist an effective treatment of Usher syndrome, and even fewer approaches for curing the disease. Nevertheless, there is more hope for Usher syndrome with these few possible treatments compared to the time when Albrecht von Graefe (1828-1870), a founder of modern ophthalmology, mentioned a connection between hearing impairment and impairment of sight for the first time over 150 years ago.
"The exceptionally slow, but obviously steady process of this disease, plus the nature of the anatomical mutation (the profound linkage of the pigment precipitation with neural atrophy), and the circumscript demarkation against the defect to a section of the visual field being in good condition and finally the frequent hereditary occurrence, all confirm the opinion this is a deep-rooted trophic defect against which therapies will stay powerless forever."
Based on current therapeutical ideas or dispositions, therapeutical prospects will hopefully not 'stay powerless forever'.
Fundamental scientific discoveries within the last couple of years are 'rays of hope'.
You can contribute, so that Usher research might lead to a fast diagnosis and a successful therapeutical solution. - Every donation counts!
Thanks!